Medical Devices: Cancer Risk Led FDA to Warn Against Certain Uses of Power Morcellators and Recommend New Labeling
Fast Facts
In 1991, FDA allowed the first laparoscopic power morcellator—a device that cuts tissue into small pieces, which can be removed during minimally invasive surgeries—on the U.S. market. In late 2013, the agency began to receive reports that these devices may spread a difficult-to-diagnose cancer when used to treat uterine fibroids.
In April 2014, FDA estimated that the risk of having the type of uterine cancer identified in these first reports was higher than previous estimates. The agency warned against the use of power morcellators to treat fibroids and recommended new labeling, which all current manufacturers followed.
Actions Taken by FDA in Response to First Adverse Event Reports about the Spread of Unsuspected Cancer Following the Use of a Laparoscopic Power Morcellator to Treat Uterine Fibroids
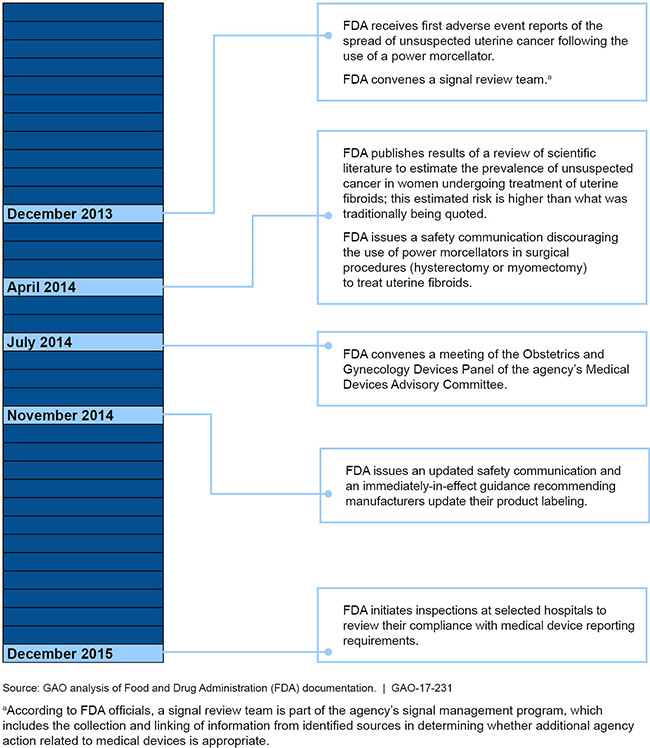
A timeline of FDA's response to reports about the spread of cancer after using a power morcellator.
Highlights
What GAO Found
Between 1991 and 2014, the Food and Drug Administration (FDA)—the federal agency responsible for the oversight of medical devices—cleared 25 submissions for laparoscopic power morcellators for the U.S. market. FDA cleared the submissions for these devices, which cut tissue into small pieces to facilitate removal through small incision sites of gynecological and other types of minimally invasive surgeries, through its premarket notification process. Under this process, established under section 510(k) of the Federal Food, Drug, and Cosmetic Act, FDA reviews information submitted by a device manufacturer and determines whether the new device is substantially equivalent to another legally marketed device, known as a predicate device. In making this determination, FDA assesses whether a device has (1) the same intended use; and (2) the same technological characteristics as a predicate device, or has different technological characteristics but submitted information demonstrates the device is as safe and effective as the predicate device, and does not raise different questions of safety or effectiveness. A device determined to be substantially equivalent is cleared to be marketed. For power morcellators, FDA determined the devices in all 25 of the 510(k) submissions had the same intended use as their predicates, while 6 had new technological characteristics.
Prior to receiving adverse event reports, FDA understood the risk of having an unsuspected cancer that could be spread using a power morcellator as low; in response to such reports, the agency has taken several actions. According to FDA officials, the agency was aware of the potential for power morcellators to spread tissue (cancerous and noncancerous) when the agency cleared the first device in 1991. FDA officials noted that, at the time, the risk of having a type of uterine cancer that can resemble noncancerous uterine tumors, called fibroids, was thought to be low based on available information. After receiving reports in December 2013 about the spread of an unsuspected cancer following the use of power morcellators in surgeries to treat fibroids, FDA estimated the cancer risk to women undergoing these surgeries to be about 1 in 350 for one type of cancer. FDA issued a safety communication in November 2014 warning against certain uses of power morcellators—specifically in treating uterine fibroids. The agency also issued guidance recommending that manufacturers add a boxed warning to their device labeling, which all current manufacturers followed, and conducted inspections to review hospitals' compliance with medical device reporting requirements. As questions remain related to the use of power morcellators, FDA has continued to monitor adverse event reports, among other actions.
Professional societies provided some guidance to physicians regarding the use of power morcellators, while manufacturers of the devices provided instructions and some technical training. According to officials at professional societies that GAO contacted, there are no professional standards specific to the use of power morcellators, but some guidance and educational resources are available for surgical procedures to treat uterine fibroids in which the devices may be used. Training requirements for physicians using power morcellators generally occur at hospitals as part of the processes to ensure that physicians have suitable experience and abilities. Manufacturers provide instructions for use, and some offer technical training that demonstrates device set-up, operation, and cleaning.
Why GAO Did This Study
In December 2013, media reports raised concerns regarding the use of power morcellators in the surgical treatment of women with uterine fibroids. These concerns focused on the spread of an unsuspected uterine cancer after such use of the devices.
GAO was asked to review power morcellator medical devices. This report examines (1) the number of 510(k) submissions for power morcellators FDA cleared, and the extent to which the agency determined the devices had new intended uses or new technological characteristics; (2) FDA's understanding of any concerns with the use of power morcellators to treat uterine fibroids prior to receiving adverse event reports, and the actions FDA has taken in response to these reports; and (3) the professional standards and guidance for physicians regarding the use of power morcellators, and the information device manufacturers provided. GAO reviewed documentation of FDA's decision-making and guidance and manufacturers' device labeling, and interviewed FDA officials. In addition, GAO reviewed documents and contacted officials from 10 professional societies and other organizations that have a potential interest in the use of power morcellators, and three health care providers that performed gynecological procedures that could involve the use of the devices. GAO also contacted all 12 manufacturers for the power morcellators FDA cleared for the U.S. market.
The Department of Health and Human Services provided technical comments on a draft of this report, which were incorporated as appropriate.
For more information, contact Marcia Crosse at (202) 512-7114 or crossem@gao.gov.